Donor Stool Product for FMT Decreases Recurrent Clostridioides difficile Infection: RBX2660 Is the First FDA-Approved Live Biotherapeutic Product

Colleen R. Kelly, MD, FACG1 and
Philip Schoenfeld, MD, MSEd, MSc (Epi)2
1Chief (Emeritus), Gastroenterology Section, John D. Dingell VA Medical Center, Detroit, MI.
2Associate Professor of Medicine, Division of Gastroenterology, Warren Alpert Medical School, Brown University, Providence, RI
3Director-IBD Center, Sibley Memorial Hospital, Washington, DC.
This summary reviews Khanna S, Assi M, Lee C et al. Efficacy and Safety of RBX2660 in PUNCH CD3, a Phase III, Randomized, Double-Blind, Placebo-Controlled Trial with a Bayesian Primary Analysis for the Prevention of Recurrent Clostridioides difficile Infection. Drugs 2022; 82: 1527–38.
Correspondence to Philip Schoenfeld, MD, MSEd, MSc. Editor-in-Cheif. Email: EBGI@gi.org
Access the article through PubMed
STRUCTURED ABSTRACT
Question: Is RBX2660 (Rebyota; Ferring Pharmaceutical, Parsippany, NJ), an enema consisting of full-spectrum donor stool microbes for fecal microbiota transplant (FMT), superior to placebo to reduce recurrent Clostridioides difficile infection (rCDI)?
Design: Phase III, multi-center, double-blind, placebo-controlled randomized controlled trial (RCT; PUNCH CD3), with a Bayesian primary analysis integrating data from a prior phase IIb RCT with similar design (PUNCH CD2).1
Setting: Forty-four sites in the US and Canada.
Patients: Included patients were > 18 years old and had rCDI (>1 recurrences after a primary CDI) or had > 2 hospitalizations with severe CDI within the past 12 months and had completed one or more courses of standard-of-care antibiotic therapy. Eligible patients were required to demonstrate a positive stool test for C. difficile toxin gene by polymerase chain reaction (PCR), enzyme immunoassay (EIA) for C. difficile toxin, or other assay within 30 days of enrollment in the trial. Multiple exclusion criteria included, but were not limited to, known history of inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), celiac disease, and prior FMT.
Interventions/Exposure: Eligible patients were randomizinfed 2:1 to RBX2660 or normal saline placebo, which was administered rectally as a single dose. The 150 ml enema was administered at the study site after the patient had completed standard-of-care antibiotic therapy plus a 24–72-hour washout period. The washout period did not include any bowel preparation prior to enema administration.
Outcome: The primary endpoint was absence of CDI diarrhea within 8 weeks of enema administration, which was defined as treatment success. The secondary endpoint was absence of CDI diarrhea within 8 weeks plus no new CDI episodes through 6 months after administration of enema, which was defined as sustained clinical response. For patients with treatment failure, open-label treatment with RBX2660 was offered.
Data Analysis: Modified intention-to-treat (mITT) analysis, defined as all randomized patients who completed treatment and 8 weeks of follow-up, was performed for the primary and secondary endpoints. In addition, a Bayesian hierarchical model* that utilized data from the dose-finding, placebo-controlled, phase IIb RCT (PUNCH CD2) was conducted. In PUNCH CD2, patients were randomized 1:1:1 to receive 2 treatment doses separated by 1 week: RBX2660 followed by another dose of RBX2660, RBX2660 followed by placebo, or placebo followed by placebo.1 Only the 1-dose RBX2660 group and the placebo group were utilized in the Bayesian analysis.
Funding: Rebiotix, a Ferring Company, and manufacturer of RBX2660.
Results: From July 2017 through February 2020, 262 patients (n = 177 for RBX2660 and n = 85 for placebo) comprised the mITT analysis: median age 63.0 (range 19-93), 68.5% female; 92.1% White; 88.0% received vancomycin alone as standard-of-care antibiotic therapy; 73.0% used PCR and 24.7% used EIA for CDI confirmation; and 36.3% had > 3 CDI episodes prior to enrollment.
In the mITT analysis for the PUNCH CD3 population, treatment success, defined as absence of CDI-associated diarrhea at 8 weeks, occurred more commonly in the RBX2660 group vs placebo: 71.2% vs 62.4%. In the PUNCH CD2 trial, 1-dose of RBX2660 (n = 44) was superior to placebo (n = 44) for treatment success (66.7% vs 45.4%, P= 0.048). Using the FDA-recommended approach to borrowing data from PUNCH CD2 and applying the Bayesian hierarchical model, the mITT analysis again showed treatment success occurred more commonly in the RBX2660 group vs placebo: 70.6% vs 57.5% with a posterior probability of success of 0.991 (i.e., 99.1% probability that RBX2660 is superior to placebo for treatment success).
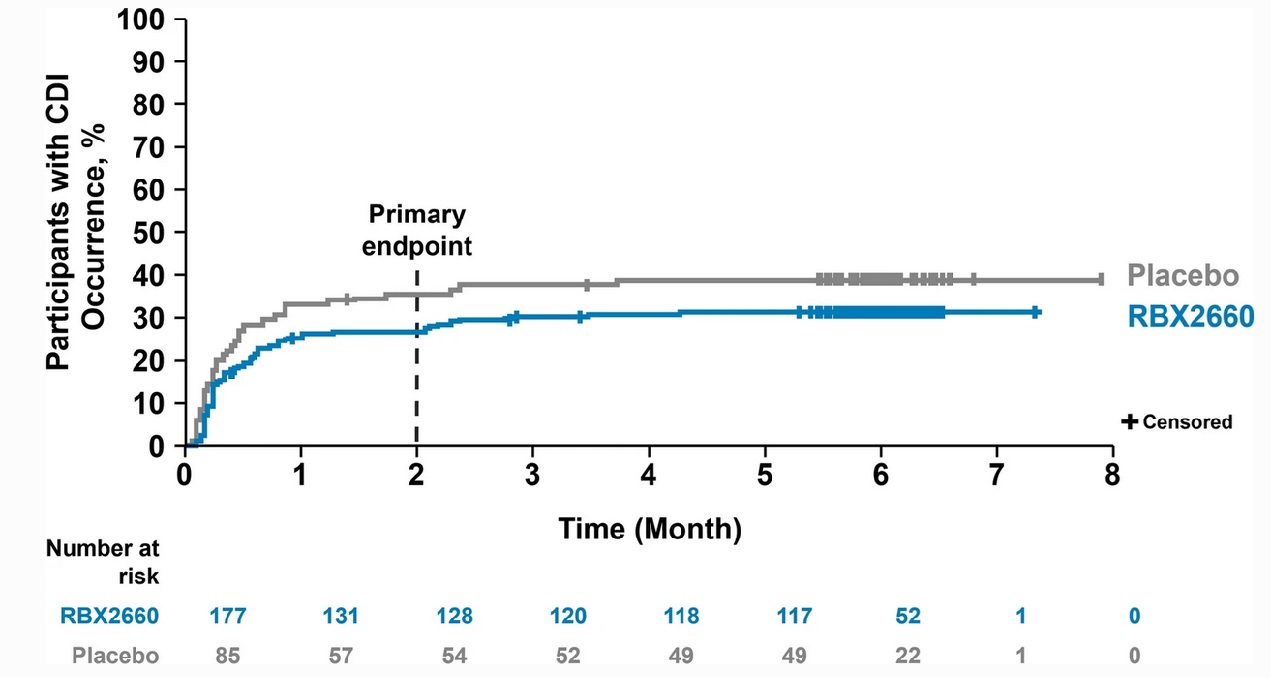
Among study patients who achieved treatment success, rCDI was infrequent at 6 months in both RBX2660 (7.9%) and placebo (8.4%) (Figure 1). Sixty-five patients had treatment failure and received open-label RBX2660. In this extension study, 62.5% of patients who had originally received placebo and 53.7% of patients who had originally received RBX 2660 achieved treatment success after getting the open-label course of treatment. No severe adverse events related to study treatment or rectal administration occurred. GI adverse events, including abdominal discomfort and diarrhea, were the only adverse events reported in more than 5% of participants in all treatment groups.
____________________________
NOTE
*Originally, 2 Phase III RCTs were planned. However, during PUNCH CD3, the study investigators noted challenges with patient recruitment (e.g., patients may be less likely to enroll in a placebo-controlled RCT when FMT for rCDI was already available as an experimental procedure under FDA’s enforcement discretion policy). After consultation with the FDA, the data analysis plan utilizing a Bayesian hierarchical model and PUNCH CD2 data in lieu of a second Phase III RCT was agreed upon. A complete discussion of Bayesian analysis is beyond the scope of this summary. The key components of Bayesian analysis are that it allows the incorporation of prior data (e.g., PUNCH CD2 data) and provides a posterior probability statement (e.g., what is the probability that RBX2660 is superior to placebo for achieving treatment success?).
COMMENTARY
Why Is This Important?
FMT has demonstrated effectiveness for the prevention of rCDI, and is currently recommended in guidelines from the American College of Gastroenterology and the Infectious Diseases Society of America.2,3 However, prior to the very recent approval of RBX2660, there were no FDA-approved FMT products and the procedure could only be performed under the agency’s policy of enforcement discretion.1,4-5 Since 2013, stool banks, such as OpenBiome, which centralize donor screening and testing to reduce risk of infection transmission, have provided most donor fecal material for FMT. However, given the FDA’s designation of donor stool as a biologic drug, there is no mechanism to regulate stool banks. The cost of donor material is not covered by insurance and patient access is limited as the supply of stool bank material is primarily reserved for centers of excellence. As of November 30, 2022, RBX2660 becomes the first FDA-approved source of donor stool for FMT to prevent rCDI.
RBX2660 contains the full spectrum of fecal microbes gathered from healthy donors. The stool is screened for multiple pathogens then processed to a stable cryopreserved liquid suspension. Per prescribing information, the cryopreserved liquid suspension is thawed and then administered as an enema6. The 150 ml enema relies on gravity for infusion of contents over 10-15 minutes with the patient in the left lateral decubitus position or a prone knee-chest position, and patients remain in position for an additional 15 minutes after the enema has been completely administered.
As discussed previously in Evidence-Based GI,4-5 the FDA is currently reviewing other live biotherapeutic products for treatment of rCDI. Specifically, SER-109 is currently under FDA review with comment due in the second quarter of 2023. SER-109 consists of capsules of donor-derived, live purified Firmicutes bacterial spores, administered orally for 3 consecutive days. In its Phase III, double-blind, placebo-controlled RCT, ECOSPOR III, it was superior to placebo for preventing rCDI: 88% vs 60%.
Key Study Findings
Caution
Although comparative RCTs are not available, FMT administered by colonoscopy and oral capsules, including SER-109, have reported treatment success in the 90% range. It’s unclear whether differences in study design or use of an enema to administer RBX2660 may have impacted treatment success rates with RBX2660. Furthermore, due to strict enrollment criteria and exclusion of patients with IBD, IBS, and other conditions, the patients enrolled in PUNCH CD3 may not represent the real-world population of patients with rCDI.
There are several study design issues that might have impacted results. Most cases of rCDI used PCR to confirm presence of C. difficile (73.0%) as opposed to requiring all study patients to have a positive EIA for C. difficile toxin. Since colonization is common and can persist for months after a CDI, some patients testing positive by PCR at the study onset may have been at low risk for rCDI and diarrhea within 8 weeks. Alternatively, during 8-week follow-up, some patients may have had functional diarrhea and false positive PCR for C. difficile and been miscategorized as treatment failure. Furthermore, no washout of antibiotics from the colon with an osmotic laxative or bowel preparation was performed prior to the enema administration of RBX2660, so it’s possible that residual antibiotics could have reduced colonization by the donor microbiota released by enema-administration of RBX2660 and decreased its efficacy.
My Practice
RBX2660 will be commercially available in the US shortly and will be the most accessible treatment option for patients with rCDI after they have completed standard-of-care antibiotic therapy and will reserve it for patients with their third episode of C. difficile (i.e., initial episode plus 2 recurrences). I’ll follow the prescribing information for administering the enema formulation of RBX2660. It is not yet clear that office-based administration of fecal enemas will be required. I’ll treat most patients after a 72-hour washout period and administer magnesium citrate one day before RBX2660 administration to minimize residual antibiotics in the colon which might reduce the efficacy of RBX2660. I anticipate the biggest challenge will be insurance coverage and cost to patients, particularly when up to 30% of patients may require repeat dosing. Stool banks operating under investigational new drug status will continue to have a role, providing donor stool for FMT in patients who cannot access commercial formulations or who fail to achieve cure after RBX2660. If SER-109 or other FDA-approved live biotherapeutic products become available, I’ll reassess my practice.
For Future Research
Given the high effectiveness of FMT and the greater availability of safe donor products, future studies should look at utilizing FMT earlier in the disease cycle, perhaps after a first or second episode, especially in patients who had a severe CDI or who are at high risk of further recurrence. Use of FMT in the acute setting to treat severe/fulminant CDI is another indication which needs further study. Real-world effectiveness in subpopulations who were excluded from industry trials, such as children and patients with IBD, will be important to understand. Finally, as new pathogens emerge, it will be important to improve and rapidly update donor screening protocols to optimize patient safety. Surveillance studies for potential safety concerns, including both infectious agents and unforeseen consequences of manipulating the gut microbiome, will continue to be important.
Conflict of Interest
Dr. Schoenfeld reports no potential conflicts of interest. Dr. Kelly reports serving as a consultant for Sebela Pharmaceuticals and is a volunteer clinical advisor for Openbiome.
@khanna_S4
@DrPaulGastro
REFERENCES
- Dubberke ER, Lee CH, Orenstein R, et al. Results from a randomized, placebo-controlled clinical trial of a RBX2660: A microbiota-based drug for the prevention of recurrent Clostridioides difficile infection. Clin Infect Dis 2018; 67: 1198-204.
- Kelly CR, Fischer M, Allegretti J, et al. ACG Clinical Guidelines: Prevention, Diagnosis, and Treatment of Clostridioides difficile Infections. Am J Gastroenterol 2021; 116: 1124-1147.
- Johnson S, Lavergne V, Skinner AM, et al. Clinical Practice Guideline by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA): 2021 Focused Update Guidelines on Management of Clostridioides difficile Infection in Adults. Clin Infect Dis 2021; 73(5): e1029-e1044.
- Allegretti JR. SER-109, an Oral Microbiome Therapy, Decreases Recurrent Clostridioides difficile Infection. Evidence-Based GI 2022; 2(4): 1-4. https://gi.org/wp-content/uploads/2022/06/Allegretti_June2022_Final-1.pdf
- Feuerstadt P, Louie TJ, Lashner B, et al. SER-109, an Oral Microbiome Therapy for Recurrent Clostridioides difficile Infection. N Engl J Med. 2022; 386(3):220-229.
- Rebyota [package insert]. Parsippany, NJ: Ferring Pharmaceuticals, 2022. https://www.ferringusa.com/pi/rebyota. Accessed December 9, 2022.
Download the article summary (PDF)